Przetwarzanie informacji jest niezbędne we wszystkich dziedzinach nauki. W biologii molekularnej centralny dogmat, po raz pierwszy ukuty przez Francisa Cricka (Crick, 1958, 1970), jest klasycznym kręgosłupem żywych komórek, który zasadniczo przeprowadza procesy od podziału komórki do śmierci poprzez szlaki informacyjne DNA, RNA i białek. Dokładniej, centralny dogmat opisuje przekazywanie informacji o sekwencji podczas replikacji DNA, transkrypcji na RNA i translacji na łańcuchy aminokwasów tworzące białka. Jednocześnie stwierdza również, że informacje nie mogą przepływać od białka do białka lub kwasu nukleinowego.
Od czasu pojawienia się podejść systemowych i wysokoprzepustowych w ciągu ostatnich dwóch dekad, te szerokie kroki, które nie obejmują skomplikowane szczegóły regulacyjne, zostały poddane intensywnej kontroli. Brakujące funkcje regulacyjne, takie jak mechanizmy korekty / naprawy DNA i alternatywny splicing pre-mRNA, wprowadzają kilka etapów pośrednich. Te dodatkowe kroki kolidują z kluczowymi krokami dogmatu i prawdopodobnie zmieniają dynamikę informacji. Ponadto epigenetyka, czyli rola odgrywana przez struktury chromatyny, metylacja DNA i modyfikacje histonów, również wydają się być sprzeczne z prostymi ścieżkami dogmatu (Shapiro, 2009; Luco i in., 2011). Splicing białek, czyli zdolność białka (intein) do zmiany własnej sekwencji, odkryta w ostatnich czasach (Volkmann i Mootz, 2012) oraz priony, które modyfikują inne sekwencje białek (Prusiner, 1998), omijają szlak transferu informacji dogmat. Inne badania wykazały błędy lub niedopasowania między sekwencjami RNA a ich kodującym DNA (Hayden, 2011; Li i wsp., 2011). Podsumowując, dane te podają w wątpliwość ważność centralnego dogmatu w kontekście współczesnej nauki, a zatem kwestionują prostotę liniowego przepływu informacji (DNA do RNA i RNA do białka).
Aby spojrzeć na sprawy z odpowiedniej perspektywy, potrzebujemy narzędzi analitycznych, które badają obawy lub rozbieżności dotyczące istniejącej od dawna teorii. Jedną z prostych, ale bardzo przydatnych technik wyszukiwania dóbr globalnych w wysokoprzepustowych zbiorach danych jest statystyczna analiza korelacji, która jest szeroko iz powodzeniem stosowana do obserwacji wzorców w złożonych systemach, takich jak pogoda (Stewart, 1990), giełdy (Lo i MacKinlay , 1988) i kosmologii (Amati i in., 2008). Istnieje kilka rodzajów analiz korelacji, które oceniają zarówno liniowe (np. Moment iloczynu Pearsona), jak i nieliniowe (np. Ranga Spearmana, wzajemne informacje) zależności (Steuer et al., 2002; Rosner, 2011). , analiza korelacji momentów iloczynu Pearsona stała się najpopularniejsza ze względu na jej zdolność do pokazania struktury organizacyjnej w najprostszej formie.
W biologii powstało wiele prac, które badały korelacje w mRNA i dane dotyczące ekspresji białek (patrz poniżej i tabela 1). Teoretycznie, gdy porównuje się dwie próbki zawierające dane wysokowymiarowe (takie jak mikromacierzy i proteomiczne), analizy korelacji dostarczają miary odchylenia od jedności jako źródła różnicy między próbkami . W skrócie, dwie próbki z identycznymi i całkowicie nieidentycznymi informacjami pokażą odpowiednio korelację jednostkową (R2 = 1) i zerową (R2 = 0).
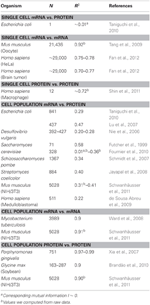
Tabela 1. mRNA i wyrażenie białka korelacje sesji w różnych organizmach.
Doskonała korelacja (R2 = 1) to sytuacja wyidealizowana, która jest daleka od rzeczywistości, ponieważ lub sam szum doświadczalny zakłóca i zmniejsza korelację. Co więcej, ostatnie lata uwypukliły istnienie szumu biologicznego: badania pojedynczych komórek i cząsteczek wykazały stochastyczność dynamiki ekspresji genów z powodu kombinatorycznego efektu niskiej liczby kopii molekularnych i kwantowego charakteru dynamiki promotora (Raj i van Oudenaarden, 2009; Eldar i Elowitz, 2010). Z drugiej strony klonalne populacje komórek wykazują heterogenność w poziomach ekspresji danego białka na komórkę w dowolnym mierzonym czasie (Chang i in., 2008). Razem stochastyczność i heterogeniczność są niezbędne do wytwarzania zróżnicowania losu komórki, zmian fenotypowych i amplifikacji sygnałów wewnątrzkomórkowych (Locke i in., 2011; Selvarajoo, 2012).
Stochastyczne fluktuacje lub wewnętrzny szum powodują ekspresja gatunków molekularnych zmienia się w czasie i między komórkami, co prowadzi do nieskorelowanych odpowiedzi (Elowitz i wsp., 2002). Jest to szczególnie widoczne w przypadku mRNA i białek o niskiej liczbie kopii. Zatem korelacja między próbkami (komórkami) może zostać obniżona z powodu wewnętrznego szumu (Rysunek 1A). Inne źródła szumu biologicznego spowodowane czynnikami zewnętrznymi obejmują zmienność wielkości komórek, liczbę kopii molekularnych i fluktuacje środowiskowe między poszczególnymi komórkami.Czynniki te zniekształcają deterministyczny centralny dogmat i prawdopodobnie zmieniają silne korelacje w słabsze (Rysunek 1B).
W jednym z ostatnich badań porównano ekspresję mRNA i białek Escherichia coli między poszczególnymi komórkami na poziomie pojedynczej cząsteczki i dostarczyło scenariusza, który głęboko kwestionuje główny dogmat. Taniguchi i in. (2010) ujawnili, że nie ma korelacji (R2 ~ 0) między poszczególnymi mRNA tufA a poziomami białka w pojedynczych komórkach. W szczególności doszli do wniosku, że brak korelacji jest prawdopodobnie spowodowany różnicami w okresach życia mRNA i białek. Chociaż jest to prawdopodobne wyjaśnienie, Taniguchi i wsp. uważali, aby nie obalić długo utrzymywanej hipotezy, twierdząc, że średnie czasowe poziomów mRNA powinny korelować z poziomami białek. Nie było jednak dowodów na to, że tak jest w rzeczywistości, a kiedy ocenialiśmy nieliniowe zależności za pomocą informacji wzajemnych (Steuer i in., 2002; Tsuchiya i in., 2010) w Taniguchi i in. zestaw danych, stwierdziliśmy, że wynik jest niezależny, tj. I ~ 0. Potwierdza to, że ekspresja mRNA do białka między poszczególnymi komórkami na poziomie pojedynczej cząsteczki jest wyraźnie niezwiązana. Ponadto podczas powiększania wykresu korelacji na poziomie pojedynczej cząsteczki jest oczywiste, że ich korelacje parami są słabe (rysunek 1A, wstawka, dla ilustracji).
Warto zauważyć, że na poziomie populacji komórek Taniguchi et glin. byli w stanie wykazać stosunkowo wysoką korelację między mRNA i ekspresją białek przy R2 = 0,29 (Rysunek 2A). W rzeczywistości, inne niezależne badanie Lu i wsp. (2007) dla populacji E. coli również wykazali stosunkowo wysoką korelację (R2 = 0,47). Podobne analizy przeprowadzone na Saccharomyces cerevisiae (Futcher i wsp., 1999), mysim fibroblastach NIH / 3T3 (Schwanhäusser i wsp., 2011) i kilku innych populacjach komórek (Nie i wsp., 2006; Schmidt i wsp., 2007; Jayapal i wsp. al., 2008; de Sousa Abreu et al., 2009) wszystkie wykazywały skorelowane struktury między ekspresją całego transkryptomu i całego proteomu (Tabela 1). Dlaczego więc nie ma korelacji między indywidualnym mRNA a ekspresją białek w pojedynczych komórkach, podczas gdy na poziomie populacji obserwuje się zbiorowe relacje między mRNA na dużą skalę a ekspresją białek?
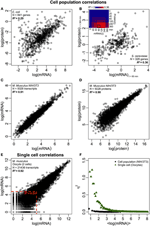
Rysunek 2. Ogólne korelacje wyrażeń omicznych. Populacje komórek: korelacje mRNA-białko w (A) E. coli (Taniguchi i wsp., 2010) i (B) S. cerevisiae (Fournier i wsp., 2010) między ekspresją mRNA w t = 60 min i ekspresją białka w t = 360 min. Wstaw: macierz korelacji między wszystkimi punktami czasowymi pokazuje opóźniony wzrost korelacji między mRNA a białkami. (C) ekspresja mRNA i (D) białek między dwiema próbkami mysich komórek NIH / 3T3 (Schwanhäusser et al., 2011). Pojedyncze komórki: (E) ekspresja mRNA między dwoma oocytami (Tang i wsp., 2009). Czerwone kropkowane linie wskazują regiony o niskiej ekspresji mRNA (log (mRNA) < 5). (F) Szum (η2) versus log (ekspresja mRNA) dla populacji komórek (NIH / 3T3, czarne kropki, Schwanhäusser et al., 2011) i pojedynczych komórek (Oocytes, zielone trójkąty, Tang et al., 2009). Każda kropka przedstawia wartość dla grupy P = 100 mRNA. η2 jest bliskie zeru dla populacji komórek dla wszystkich ekspresji mRNA. W przypadku pojedynczych komórek η2 jest najwyższe dla mRNA o najniższej liczbie kopii i zbliża się do zera dla większej liczby kopii.
Uważamy, że istnieją dwa główne powody różnic. Po pierwsze, jak wspomniano wcześniej, hałas, zarówno biologiczny, jak i niebiologiczny, zmniejsza korelację. Ponieważ analizy pojedynczych komórek wykazały znaczenie stochastyczności i zmienności, efekty te mają kluczowe znaczenie dla redukcji korelacji pojedynczych komórek. Na poziomie zespołu, gdy komórki są pobierane do populacji, całkowity szum (wewnętrzny + zewnętrzny) jest zredukowany, ponieważ szum losowy jest eliminowany we wszystkich zakresach ekspresji molekularnych (rysunki 1C – F), aby ujawnić średnią odpowiedź i samoorganizację ( Karsenti, 2008; Selvarajoo, 2011; Hekstra i Leibler, 2012; Selvarajoo i Giuliani, 2012). Stąd, pojawia się dobry stopień korelacji ekspresji mRNA-białko. Po drugie, w badaniu pojedynczej komórki (Taniguchi i wsp., 2010) porównano indywidualną korelację ekspresji mRNA-białko w wielu komórkach. Jednak w badaniach populacji komórek porównuje się je w całości, na tysiącach mRNA i białek o kilka rzędów wielkości większych niż zakres ekspresji stwierdzony dla pojedynczej cząsteczki między komórkami. W związku z tym prowadzi to do wyższych korelacji na poziomie populacji, ponieważ wpływ pojedynczych zmian molekularnych staje się nieistotny.
Pomimo obserwowanych skorelowanych struktur dla populacji komórek, istnieją namacalne przyczyny dużego odchylenia od doskonałej korelacji.Jak wspomniano wcześniej, kluczową kwestią jest to, że mRNA i białka są zlokalizowane sekwencyjnie z kilkoma brakującymi procesami, które nie są reprezentowane w centralnym dogmacie. Dodanie brakujących półproduktów do ścieżki biochemicznej spowoduje zauważalne opóźnienie w przepływie informacji (Selvarajoo, 2006, 2011; Piras i in., 2011), a korelacja między nimi może ucierpieć. Może to być również częścią faktu odnotowanego przez Taniguchi i wsp. że ekspresje mRNA i białek mają różne okresy życia. Warto zauważyć, że postulat ten jest poparty w niedawnej pracy dotyczącej S. cerevisiae leczonej Rapamycyną, która wykazała, że czasowe korelacje ekspresji mRNA-białka były początkowo niskie, R2 = 0,01 po 40 min, niemniej jednak po 360 min po zaburzeniu korelacja wzrosła, R2 = 0,36 (Fournier i in., 2010, Rysunek 2B). Dane wskazują, że po zaburzeniu chemicznym początkowa odpowiedź między mRNA a ekspresją białek jest odchylona z powodu opóźnienia czasowego i różnych mechanizmów kinetycznych między nimi, a także efektów wtórnych, takich jak autokrynne lub parakrynne zakłócenia sygnalizacji (Shvartsman i in., 2002; Isalan i in., 2008). Kiedy efekty zaburzeń są osłabione w czasie, nastąpiło przywrócenie korelacji.
Aby dodatkowo sprawdzić postulację, że sekwencyjne procesy opóźnień lub różne okresy życia są kluczowe dla zmniejszenia korelacji mRNA-białko, porównaliśmy R2 między ten sam gatunek molekularny centralnego dogmatu (np. między mRNA i mRNA) w populacjach komórek i pojedynczych komórkach. Korelacja ekspresji mRNA-mRNA w całym transkryptomie między powtórzeniami NIH / 3T3 (Schwanhäusser et al., 2011) (Rysunek 2C) i Mycobacterium tuberculosis (Ward et al., 2008) jest bardzo wysoka, z R2 > 0.9 (Tabela 1). Takie silne korelacje obserwuje się również między próbkami populacji pod kątem ekspresji białko-białko w komórkach NIH / 3T3 (Schwanhäusser i in., 2011) (Rysunek 2D), Porphyromonas gingivalis (Xia i wsp., 2007) i Glycine max (Brandão i in. , 2010) (tabela 1). Ponieważ te dane, które porównują te same gatunki, dają bardzo wysokie korelacje, można sobie wyobrazić, że sekwencyjne procesy opóźnienia lub różne okresy życia są odpowiedzialne za obniżenie struktur korelacji poziomu populacji między ekspresją mRNA i białek.
W pojedynczych mysich oocytach ( Tang i in., 2009), porównując całą ekspresję mRNA – mRNA, obserwuje się silnie skorelowaną strukturę (R2 = 0,92, Rysunek 2E). Jednak skupiając się tylko na mRNA o niskiej ekspresji (z wyrażeniami logarytmicznymi < 5), szum stochastyczny dość dramatycznie obniża korelację par (R2 < 0,54). Aby zbadać ten wynik, oceniliśmy szum, η2 = σ2XY / μ2XY, w całej ekspresji mRNA (Rysunek 2F). Zauważyliśmy, że η2 jest najwyższe dla najniższych wyrażeń, ze względu na wyraźny efekt fluktuacji stochastycznych w porównaniu z ich wyrażeniami i zbliża się do zera dla wyższych wyrażeń, gdzie taki szum staje się mniej istotny (Piras i in., 2012). W przypadku populacji komórek, zgodnie z oczekiwaniami, w całym zakresie ekspresji obserwuje się szum prawie zerowy ze względu na eliminację szumu losowego (ryc. 1E, F).
Silnie skorelowane struktury dla całej ekspresji mRNA – mRNA również zgłoszono dla pojedynczej komórki rakowej (Fan i in., 2012), chociaż mniej istotne przy R2 ~ 0,7 (Tabela 1). Ponadto porównanie ekspresji białko-białko w stymulowanych LPS ludzkich makrofagach również wykazało wysokie korelacje, R2 ~ 0,72 (Shin i wsp., 2011) (Tabela 1). Chociaż nie ma korelacji między ekspresją poszczególnych mRNA-białek w pojedynczych komórkach, korelacja na dużą skalę lub w skali omicznej między tymi samymi gatunkami molekularnymi w pojedynczych komórkach jest bardzo wysoka.
Zatem czy pojedyncze komórki czy populacje komórek , dane omiczne wskazują, że korelacje między tymi samymi gatunkami molekularnymi (mRNA vs. mRNA i białko vs. białko) są zauważalnie wyższe niż między różnymi gatunkami (mRNA vs. białko). Odzwierciedla to fakt, że chociaż procesy opóźnienia czasowego i różne okresy życia są kluczowe dla zmniejszenia korelacji, mechanizmy te nie są wystarczające do wspierania braku struktury korelacji obserwowanej między indywidualnymi transkryptami a ekspresjami białek pojedynczych komórek.
A więc do tej pory, badając ekspresję mRNA i białek różnych systemów komórkowych na dużą skalę, wykazaliśmy, że struktury korelacyjne wyłaniają się w skali globalnej, jednak analizy korelacji ujawniają jedynie łączność między dwiema badanymi próbkami i nie pokazują kierunku przepływ informacji. Aby centralny dogmat obowiązywał w skali globalnej, ogólny przepływ informacji powinien przebiegać od DNA do białek. Taki przepływ informacji został wykazany w niezliczonych innych badaniach, które obejmują zakłócanie receptorów populacji komórek i monitorowanie wynikowego dynamika czynników transkrypcyjnych wiążących się z DNA i indukcja ekspresji genów na dużą skalę (ryc. 3A).Na przykład w przypadku komórek odpornościowych stymulowanych LPS wykazano, że aktywacja czynnika transkrypcyjnego NF-κB następuje po około 15 minutach (Liu i wsp., 1999), a indukcja jego dalszych genów przy około 30 minutach min (Liu i wsp., 1999; Xaus i wsp., 2000; Selvarajoo i wsp., 2008) oraz translację odpowiednich białek w zakresie 60-90 min (Kawai i wsp., 1999; Xaus i wsp. ., 2000) (Rysunek 3B). Taki sekwencyjny kierunek całego przepływu informacji transkrypcji na translację obserwuje się również w przypadku systemów bakteryjnych, takich jak E. coli, na poziomie populacji komórek (Golding i wsp., 2005).

Rysunek 3. Przepływ informacji w centralnym dogmacie. (A) Schemat ekspresji TNF indukowanej przez LPS / TLR4, poprzez czynnik transkrypcyjny NF-κB i gen tnf, po liniowym przepływie informacji. (B) Doświadczalne profile czasowe aktywności wiązania promotora ekspresji NF-κB (panele górne), tnf (panele środkowe) i TNF (panele dolne) na poziomie populacji komórek. (C) Schematyczne profile czasowe dynamiki promotora, mRNA i ekspresji białek na poziomie pojedynczej komórki (Raj i van Oudenaarden, 2009).
Alternatywnie, badania przy rozdzielczości pojedynczej komórki ujawniają losowe fluktuacje w liniowym przepływie informacji: czynniki transkrypcyjne wiążące się z regionami promotora DNA mają charakter ilościowy, co skutkuje pękaniem transkrypcji mRNA, a następnie indukuje zmienność w translacji białek, nawet między identycznymi komórkami (Rysunek 3C) (Raj i van Oudenaarden, 2009; Eldar i Elowitz, 2010; Locke i in., 2011; Hekstra i Leibler, 2012; Selvarajoo, 2012). W rezultacie w dowolnym momencie indywidualna odpowiedź molekularna pojedynczych komórek jest raczej hałaśliwa w porównaniu ze średnią skalą populacji (Selvarajoo, 2011).
Wnioski
Przykłady pokazane w W artykule podkreślono różnice w kolejności wartości korelacji obserwowanych między gatunkami w centralnym dogmacie w stosunku do populacji komórek i pojedynczych komórek. Analizy statystyczne populacji komórek dają obraz, że korelacja ekspresji między tymi samymi gatunkami molekularnymi jest bardzo wysoka, a między gatunkami umiarkowanie wysoka. Chociaż korelacje pojedynczych komórek między tymi samymi gatunkami są porównywalne z populacjami komórek, wykazywały one szerszy rozrzut na wykresach ekspresji ze względu na wyraźny efekt szumu biologicznego, szczególnie w przypadku transkryptów o niskiej liczbie kopii. Warto zauważyć, że korelacja parami pojedynczych komórek staje się zerowa dla poszczególnych cząsteczek (Taniguchi i in., 2010). W rzeczywistości wiadomo, że fluktuacje stochastyczne i zmienność ekspresji molekularnych są funkcjonalne w generowaniu decyzji o losie komórki i przechylaniu stanów komórkowych (Losick i Desplan, 2008; Eldar i Elowitz, 2010; Kuwahara i Schwartz, 2012). Uważamy, że silne korelacje omiczne występują w wyniku ścisłych sieci regulatorowych genów i białek w tysiącach cząsteczek (Barabási i Oltvai, 2004; Karsenti , 2008), w wyniku czego pojawiają się średnie odpowiedzi. Analizując niewielką liczbę lub pojedyncze cząsteczki, nie można zaobserwować struktury korelacji.
Ogólnie można sobie wyobrazić, że oglądanie przepływu informacji pojedynczego DNA do białka podważy centralny dogmat ponieważ reakcja każdej cząsteczki w dowolnym momencie prawdopodobnie nie będzie korelować. Jednak globalnie obserwacja średniej deterministycznej odpowiedzi sugeruje, że równowaga netto genów Informacje ic pozostają po prawej stronie ścieżek. Dlatego centralny dogmat należy postrzegać jako makroskopowy przepływ informacji komórkowej na skalę omiczną, a nie na poziomie pojedynczego genu do białka. W związku z tym uważamy, że jego prostota pozostanie jednym z najbardziej wpływowych teoretycznych filarów systemów żywych.
Oświadczenie o konflikcie interesów
Autorzy deklarują, że badanie zostało przeprowadzone w brak jakichkolwiek relacji handlowych lub finansowych, które mogłyby zostać zinterpretowane jako potencjalny konflikt interesów.
Podziękowania
Kentaro Hayashi jest wdzięczny za komentarze. Fundusz badawczy miasta Tsuruoka i prefektury Yamagata jest doceniany za ich wsparcie.
Crick, F. (1958). O syntezie białek. Symp. Soc. Exp. Biol. 12, 139–163.
Pubmed Abstract | Pubmed Full Text
Crick, F. (1970). Centralny dogmat biologii molekularnej. Nature 227, 561–563.
Pubmed Abstract | Publikowany pełny tekst
Hayden, E. C. (2011). Dowody na zmieniony RNA wywołują dyskusję. Nature 473, 432.
Pubmed Abstract | Pubmed pełny tekst | CrossRef Full Text
Hekstra, D. R. i Leibler, S. (2012). Przepisy awaryjne i statystyczne w replikowanych mikrobiologicznych zamkniętych ekosystemach. Cell 149, 1164–1173.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Kuwahara, H. i Schwartz, R. (2012).Stochastyczny zysk w stanie równowagi w procesie ekspresji genów z kontrolą degradacji mRNA. J. R. Soc. Interface 9, 1589–1598.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Nie, L., Wu, G. i Zhang, W. (2006). Korelacja ekspresji mRNA i obfitości białek, na którą wpływają cechy wielu sekwencji związanych z wydajnością translacji w Desulfovibrio vulgaris: analiza ilościowa. Genetics 174, 2229–2243.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Prusiner, S. B. (1998). Priony. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Rosner, B. (2011). Podstawy biostatystyki. 7th Edn. Boston, MA: Duxbury Press.
Selvarajoo, K. (2006). Odkrywanie mechanizmu aktywacji różnicowej szlaków sygnałowych receptora Toll-podobnego 4 w nokautach MyD88. FEBS Lett. 580, 1457–1464.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Selvarajoo, K. (2011). Makroskopowe prawo zachowania ujawniło się w dynamice populacyjnej sygnalizacji receptorów typu toll-like. Cell Commun. Sygnał. 9, 9.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Selvarajoo, K. (2012). Zrozumienie multimodalnych decyzji biologicznych na podstawie dynamiki pojedynczej komórki i populacji. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 385–399.
Pubmed Abstract | Pubmed pełny tekst | Pełny tekst CrossRef
Stewart, T. R. (1990). Dekompozycja współczynnika korelacji i wykorzystanie go w analizie umiejętności prognozowania. Prognoza pogody. 5, 661–666.