Lelaborazione delle informazioni è essenziale in tutti i campi della scienza. Nella biologia molecolare, il dogma centrale, coniato per la prima volta da Francis Crick (Crick, 1958, 1970), è una spina dorsale classica delle cellule viventi per eseguire fondamentalmente i processi dalla divisione cellulare alla morte attraverso i percorsi di informazione del DNA, dellRNA e delle proteine. Più specificamente, il dogma centrale descrive il trasferimento di informazioni di sequenza durante la replicazione del DNA, la trascrizione in RNA e la traduzione in catene di amminoacidi che formano proteine. Allo stesso tempo, afferma anche che le informazioni non possono fluire dalle proteine alle proteine o agli acidi nucleici.
Dallavvento di approcci sistemici e ad alto rendimento negli ultimi due decenni, questi ampi passaggi, che non includono dettagli normativi complessi, sono stati oggetto di un attento esame. Le caratteristiche normative mancanti, come i meccanismi di correzione / riparazione del DNA e lo splicing alternativo del pre-mRNA, introducono diversi passaggi intermedi. Questi passaggi aggiuntivi interferiscono con i passaggi chiave del dogma e probabilmente alterano la dinamica dellinformazione. Inoltre, anche lepigenetica, o il ruolo svolto dalle strutture della cromatina, dalla metilazione del DNA e dalle modificazioni dellistone, sembrano andare contro i semplici percorsi del dogma (Shapiro, 2009; Luco et al., 2011). Lo splicing proteico, o la capacità di una proteina (inteins) di alterare la propria sequenza, scoperto in tempi recenti (Volkmann e Mootz, 2012) e prioni, che modificano altre sequenze proteiche (Prusiner, 1998), aggirano il percorso di trasferimento delle informazioni del dogma. Altre indagini hanno riportato errori o discrepanze tra le sequenze di RNA e il loro DNA codificante (Hayden, 2011; Li et al., 2011). Presi insieme, questi dati sollevano dubbi sulla validità del dogma centrale nel contesto della scienza odierna e, quindi, mettono in dubbio la semplicità del flusso di informazioni lineari (da DNA a RNA e da RNA a proteine).
Per mettere le cose in prospettiva, abbiamo bisogno di strumenti analitici che indagano le preoccupazioni o le discrepanze riguardanti la teoria di vecchia data. Una tecnica semplice, ma molto utile per la ricerca di proprietà globali in set di dati ad alto rendimento è lanalisi di correlazione statistica, che è stata ampiamente e con successo utilizzata per osservare modelli in sistemi complessi come il tempo (Stewart, 1990), i mercati azionari (Lo e MacKinlay , 1988) e cosmologia (Amati et al., 2008). Esistono diversi tipi di analisi di correlazione che valutano le dipendenze sia lineari (es. Momento prodotto di Pearson) che non lineari (es. Rango di Spearman, informazione reciproca) (Steuer et al., 2002; Rosner, 2011). , lanalisi di correlazione prodotto-momento di Pearson è diventata la più popolare grazie alla sua capacità di mostrare la struttura organizzativa nella forma più semplice.
In biologia, ci sono stati numerosi lavori che hanno studiato le correlazioni nellmRNA e dati di espressione proteica (vedere sotto e Tabella 1). In teoria, quando vengono confrontati due campioni contenenti dati ad alta dimensione (come microarray e proteomica), le analisi di correlazione forniscono una misura della deviazione dallunità come fonte di differenza tra i campioni In breve, due campioni con informazioni identiche e completamente non identiche mostreranno la correlazione di unità (R2 = 1) e nulla (R2 = 0), rispettivamente.
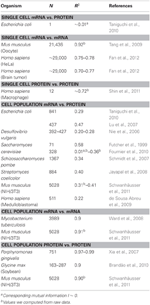
Tabella 1. mRNA ed espressione di proteine correlazioni di essione in vari organismi.
La correlazione perfetta (R2 = 1) è una situazione idealizzata che è lontana dalla realtà, in quanto tecnica o il rumore sperimentale da solo interferisce e riduce la correlazione. Inoltre, gli ultimi anni hanno evidenziato lesistenza del rumore biologico: gli studi su singole cellule e molecole hanno dimostrato la stocasticità nelle dinamiche di espressione genica dovuta alleffetto combinatorio di bassi numeri di copie molecolari e alla natura quantale della dinamica del promotore (Raj e van Oudenaarden, 2009; Eldar e Elowitz, 2010). Daltra parte, le popolazioni clonali di cellule mostrano eterogeneità nei livelli di una data espressione proteica per cellula in qualsiasi momento misurato (Chang et al., 2008). Insieme, la stocasticità e leterogeneità sono essenziali per produrre diversificazione del destino cellulare, variazioni fenotipiche e amplificazione dei segnali intracellulari (Locke et al., 2011; Selvarajoo, 2012).
Le fluttuazioni stocastiche, o rumore intrinseco, causano lespressione di una specie molecolare che varia nel tempo e tra le cellule, portando a risposte non correlate (Elowitz et al., 2002). Ciò è particolarmente importante per mRNA e proteine con un numero di copie basso. Pertanto, la correlazione tra campioni (cellule) può essere ridotta a causa del rumore intrinseco (Figura 1A). Altre fonti di rumore biologico dovuto a fattori estrinseci includono la variabilità nelle dimensioni delle cellule, il numero di copie molecolari e le fluttuazioni ambientali tra le singole cellule.Questi fattori distorcono il dogma deterministico centrale e probabilmente alterano le correlazioni forti in quelle più deboli (Figura 1B).
Uno studio recente ha confrontato lmRNA di Escherichia coli e le espressioni proteiche tra singole cellule a livello di singola molecola e ha fornito uno scenario che mette in discussione profondamente il dogma centrale. Taniguchi et al. (2010) hanno rivelato che non esiste alcuna correlazione (R2 ~ 0) tra lmRNA di tufA individuale e i livelli di proteine nelle singole cellule. In particolare, hanno concluso che la mancanza di correlazione è probabilmente dovuta a differenze nella durata di mRNA e proteine. Sebbene questa sia una spiegazione plausibile, Taniguchi et al. sono stati attenti a non confutare lipotesi di lunga durata sostenendo che le medie temporali dei livelli di mRNA dovrebbero essere correlate ai livelli di proteine. Tuttavia, non è stata dimostrata alcuna prova per dimostrare che questo è il caso reale, e quando abbiamo valutato le dipendenze non lineari utilizzando informazioni reciproche (Steuer et al., 2002; Tsuchiya et al., 2010) in Taniguchi et al. set di dati, abbiamo trovato il risultato non dipendente, cioè I ~ 0. Ciò conferma che le espressioni di mRNA e proteine tra singole cellule a livello di singola molecola sono chiaramente non correlate. Inoltre, quando si esegue lo zoom a livello di singola molecola nel grafico di correlazione, è evidente che le loro correlazioni a coppie sono deboli (Figura 1A, inserire, per lillustrazione).
In particolare, a livello di popolazione cellulare, Taniguchi et al. sono stati in grado di mostrare una correlazione relativamente alta tra mRNA e espressioni proteiche con R2 = 0,29 (Figura 2A). In effetti, un altro studio indipendente di Lu et al. (2007), per la popolazione di E. coli, ha anche mostrato una correlazione relativamente alta (R2 = 0,47). Analisi simili eseguite su Saccharomyces cerevisiae (Futcher et al., 1999), fibroblasto murino NIH / 3T3 (Schwanhäusser et al., 2011) e diverse altre popolazioni cellulari (Nie et al., 2006; Schmidt et al., 2007; Jayapal et al. al., 2008; de Sousa Abreu et al., 2009) hanno tutti mostrato strutture correlate tra espressioni a livello di trascrittoma e a livello di proteoma (Tabella 1). Allora, perché non cè correlazione tra i singoli mRNA e le espressioni proteiche nelle singole cellule, mentre a livello di popolazione si osservano relazioni collettive tra lmRNA su larga scala e le espressioni proteiche?
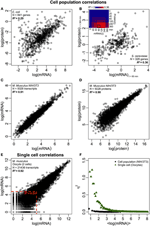
Figura 2. Correlazioni di espressioni omiche. Popolazioni cellulari: correlazioni mRNA-proteina in (A) E. coli (Taniguchi et al., 2010) e (B) S. cerevisiae (Fournier et al., 2010) tra espressioni di mRNA at = 60 min ed espressioni proteiche at = 360 min. Inserto: la matrice di correlazione tra tutti i punti temporali mostra un aumento ritardato delle correlazioni tra mRNA e proteine. (C) espressioni di mRNA e (D) proteine tra due campioni di cellule murine NIH / 3T3 (Schwanhäusser et al., 2011). Cellule singole: (E) espressioni di mRNA tra due ovociti (Tang et al., 2009). Le linee tratteggiate rosse indicano le regioni di espressioni a basso mRNA (log (mRNA) < 5). (F) Rumore (η2) rispetto a log (espressioni di mRNA) per la popolazione cellulare (NIH / 3T3, punti neri, Schwanhäusser et al., 2011) e singole cellule (ovociti, triangoli verdi, Tang et al., 2009). Ogni punto rappresenta il valore per un gruppo di P = 100 mRNA. η2 è vicino allo zero per la popolazione cellulare per tutte le espressioni di mRNA. Per le singole cellule, η2 è il più alto per gli mRNA con il numero di copie più basso e si avvicina a zero per i numeri di copie più alti.
Crediamo ci sono due ragioni principali per le differenze. In primo luogo, come notato in precedenza, il rumore, sia di natura biologica che non biologica, riduce la correlazione. Poiché le analisi su singole cellule hanno dimostrato limportanza della stocasticità e della variabilità, questi effetti sono cruciali per ridurre le correlazioni delle singole cellule. A livello di insieme, quando le cellule vengono campionate in una popolazione, il rumore totale (intrinseco + estrinseco) viene ridotto, poiché il rumore casuale si annulla in tutta la gamma di espressioni molecolari (Figure 1C-F), per rivelare la risposta media e lauto-organizzazione ( Karsenti, 2008; Selvarajoo, 2011; Hekstra e Leibler, 2012; Selvarajoo e Giuliani, 2012). Quindi, emerge un buon grado di correlazione di espressione di mRNA-proteina. In secondo luogo, per lo studio su cellula singola (Taniguchi et al., 2010), è stata confrontata la correlazione di espressione di mRNA-proteina individuale tra numerose cellule. Negli studi sulla popolazione cellulare, tuttavia, il confronto viene effettuato nella sua interezza, tra migliaia di mRNA e proteine su diversi ordini di grandezza maggiore dellintervallo di espressione trovato per una singola molecola tra le cellule. Ciò, quindi, porta a correlazioni più elevate a livello di popolazione poiché leffetto delle singole variazioni molecolari diventa trascurabile.
Nonostante le strutture correlate siano osservate per le popolazioni cellulari, ci sono ragioni tangibili per la grande deviazione dalla correlazione perfetta.Come notato in precedenza, un punto chiave è che gli mRNA e le proteine sono localizzati in sequenza con diversi processi mancanti, non rappresentati nel dogma centrale. Laggiunta degli intermedi mancanti lungo un percorso biochimico comporterà un notevole ritardo nel flusso di informazioni (Selvarajoo, 2006, 2011; Piras et al., 2011) e la correlazione tra loro potrebbe risentirne. Questo potrebbe anche essere parte del fatto osservato da Taniguchi et al. che le espressioni di mRNA e proteine hanno vite diverse. In particolare, questa postulazione è supportata in un recente lavoro su S. cerevisiae trattato con Rapamicina che ha mostrato che le correlazioni temporali dellespressione della proteina mRNA erano inizialmente basse, R2 = 0,01 a 40 min, tuttavia, oltre 360 min dopo la perturbazione, la correlazione è aumentata, R2 = 0,36 (Fournier et al., 2010, Figura 2B). I dati indicano che in seguito a perturbazioni chimiche, la risposta iniziale tra le espressioni di mRNA e proteine devia a causa del ritardo temporale e dei diversi meccanismi cinetici tra di loro, così come per gli effetti secondari come linterferenza di segnalazione autocrina o paracrina (Shvartsman et al., 2002; Isalan et al., 2008). Quando gli effetti della perturbazione si attenuano nel tempo, si è verificato il recupero delle correlazioni.
Per verificare ulteriormente la postulazione che i processi di ritardo sequenziali o le diverse vite sono cruciali per diminuire le correlazioni mRNA-proteina, abbiamo confrontato R2 tra i stesse specie molecolari del dogma centrale (ad esempio, tra mRNA e mRNA) in popolazioni cellulari e singole cellule. La correlazione dellespressione di mRNA-mRNA a livello di trascrittoma tra i replicati di NIH / 3T3 (Schwanhäusser et al., 2011) (Figura 2C) e Mycobacterium tuberculosis (Ward et al., 2008) è molto alta, con R2 > 0,9 (Tabella 1). Tali forti correlazioni si osservano anche tra campioni di popolazione per le espressioni proteina-proteina nelle cellule NIH / 3T3 (Schwanhäusser et al., 2011) (Figura 2D), Porphyromonas gingivalis (Xia et al., 2007) e Glycine max (Brandão et al. , 2010) (Tabella 1). Poiché questi dati che confrontano le stesse specie producono correlazioni molto elevate, è concepibile che i processi di ritardo sequenziale o le diverse vite siano responsabili dellabbassamento delle strutture di correlazione a livello di popolazione tra le espressioni di mRNA e proteine.
In singoli ovociti murini ( Tang et al., 2009), quando si confrontano intere espressioni di mRNA-mRNA, si osserva una struttura altamente correlata (R2 = 0,92, Figura 2E). Tuttavia, concentrandosi solo sugli mRNA poco espressi (con espressioni logaritmiche < 5), il rumore stocastico abbassa la correlazione a coppie in modo abbastanza drammatico (R2 < 0,54). Per sondare questo risultato abbiamo valutato il rumore, η2 = σ2XY / μ2XY, attraverso intere espressioni di mRNA (Figura 2F). Abbiamo notato che η2 è più alto per le espressioni più basse, a causa delleffetto pronunciato delle fluttuazioni stocastiche rispetto alle loro espressioni, e si avvicina a zero per le espressioni più alte, dove tale rumore diventa meno significativo (Piras et al., 2012). Per la popolazione cellulare, come previsto, si osserva un rumore vicino allo zero nellintero intervallo di espressione a causa dellannullamento del rumore casuale (Figure 1E, F).
Sono state anche osservate strutture altamente correlate per intere espressioni di mRNA-mRNA riportato per singola cellula tumorale (Fan et al., 2012), anche se meno significativo con R2 ~ 0,7 (Tabella 1). Inoltre, il confronto delle espressioni proteina-proteina nei macrofagi umani stimolati con LPS ha mostrato anche correlazioni elevate, R2 ~ 0,72 (Shin et al., 2011) (Tabella 1). Sebbene non vi sia correlazione tra le singole espressioni di mRNA-proteina in singole cellule, la correlazione su larga scala o omica tra le stesse specie molecolari in singole cellule è molto alta.
Pertanto, sia che si tratti di singole cellule o di popolazioni cellulari , i dati su scala omica indicano che le correlazioni tra le stesse specie molecolari (mRNA vs mRNA e proteina vs proteina) sono notevolmente più alte rispetto a specie diverse (mRNA vs proteina). Ciò riflette il fatto che, sebbene i processi di ritardo nel tempo e le diverse durate di vita siano fondamentali per ridurre le correlazioni, questi meccanismi non sono sufficienti per supportare la mancanza di struttura di correlazione osservata tra la trascrizione individuale delle singole cellule nelle espressioni proteiche.
Quindi fino ad ora, attraverso lo studio di espressioni su larga scala di mRNA e proteine di vari sistemi cellulari, abbiamo dimostrato che le strutture di correlazione emergono su scala globale. Tuttavia, le analisi di correlazione rivelano solo la connettività tra due campioni testati e non mostrano la direzione di flusso di informazioni. Affinché il dogma centrale sia valido su scala globale, il flusso complessivo di informazioni dovrebbe essere dal DNA alle proteine. Tale flusso di informazioni è stato dimostrato da una miriade di altri studi che coinvolgono perturbare i recettori delle popolazioni cellulari e monitorare la risultante dinamica dei fattori di trascrizione che si legano al DNA e linduzione di espressioni geniche su larga scala (Figura 3A).Ad esempio, nel caso di cellule immunitarie stimolate da LPS, è stato dimostrato che lattivazione del fattore di trascrizione NF-κB avviene a circa 15 min (Liu et al., 1999), linduzione dei suoi geni a valle a circa 30 min (Liu et al., 1999; Xaus et al., 2000; Selvarajoo et al., 2008) e la traduzione delle corrispondenti proteine nella regione di 60-90 min (Kawai et al., 1999; Xaus et al. ., 2000) (Figura 3B). Tale direzione sequenziale della trascrizione globale al flusso di informazioni di traduzione si osserva anche per sistemi batterici, come E. coli, a livello di popolazione cellulare (Golding et al., 2005).

Figura 3. Il flusso di informazioni del dogma centrale. (A) Schema dellespressione TNF indotta da LPS / TLR4, tramite il fattore di trascrizione NF-κB e il gene tnf, seguendo il flusso di informazioni lineari. (B) Profili temporali sperimentali dellattività di legame del promotore delle espressioni NF-κB (pannelli superiori), tnf (pannelli centrali) e TNF (pannelli inferiori) a livello di popolazione cellulare. (C) Profili temporali schematici di dinamiche del promotore, mRNA ed espressioni di proteine a livello di singola cellula (Raj e van Oudenaarden, 2009).
In alternativa, le indagini sulla risoluzione di una singola cellula rivelano fluttuazioni casuali nel flusso di informazioni lineari: i fattori di trascrizione che si legano alle regioni del promotore del DNA sono quantali, determinando un comportamento esplosivo della trascrizione dellmRNA e, successivamente, induce variabilità nella traduzione della proteina, anche tra cellule identiche (Figura 3C) (Raj e van Oudenaarden, 2009; Eldar ed Elowitz, 2010; Locke et al., 2011; Hekstra e Leibler, 2012; Selvarajoo, 2012). Di conseguenza, in qualsiasi momento particolare, la risposta molecolare individuale per singole cellule è piuttosto rumorosa rispetto alla scala media della popolazione (Selvarajoo, 2011).
Conclusioni
Gli esempi mostrati in questo documento evidenzia le differenze nellordine dei valori di correlazione osservati tra le specie nel dogma centrale rispetto alle popolazioni cellulari e alle singole cellule. Le analisi statistiche delle popolazioni cellulari dipingono un quadro che la correlazione di espressione tra le stesse specie molecolari è molto alta e tra le specie è moderatamente alta. Sebbene le correlazioni di singole cellule tra le stesse specie siano paragonabili alle popolazioni cellulari, hanno mostrato una dispersione più ampia nei loro grafici di espressione a causa delleffetto pronunciato del rumore biologico, specialmente per le trascrizioni con un numero di copie basso. In particolare, la correlazione a coppie delle singole cellule diventa zero per le singole molecole (Taniguchi et al., 2010). Infatti, le fluttuazioni stocastiche e la variabilità nelle espressioni molecolari sono note per essere funzionali nel generare decisioni sul destino cellulare e nel ribaltare gli stati cellulari (Losick e Desplan, 2008; Eldar e Elowitz, 2010; Kuwahara e Schwartz, 2012). Riteniamo che le forti correlazioni omiche si verifichino come risultato di strette reti di regolazione genica e proteica attraverso migliaia di molecole (Barabási e Oltvai, 2004; Karsenti , 2008) con conseguente risposte medie emergenti. Analizzando un piccolo numero o singole molecole, la struttura di correlazione non può essere osservata.
Nel complesso, è concepibile che la visualizzazione del flusso di informazioni dal singolo DNA alla proteina metta in discussione il dogma centrale poiché la risposta di ciascuna molecola in un singolo momento non sarà probabilmente correlata. Tuttavia, a livello globale, losservazione della risposta deterministica media suggerisce che lequilibrio netto della genetta le informazioni IC rimangono allestrema destra dei percorsi. Pertanto, il dogma centrale dovrebbe essere visto come un flusso di informazioni cellulari macroscopiche su scala omica e non a livello di singolo gene a livello di proteina. In quanto tale, riteniamo che la sua semplicità continuerà a rimanere uno dei pilastri teorici più influenti dei sistemi viventi.
Conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in lassenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Ringraziamenti
Kentaro Hayashi è ringraziato per i commenti. Il fondo di ricerca della città di Tsuruoka e della prefettura di Yamagata è apprezzato per il loro sostegno.
Crick, F. (1958). Sulla sintesi proteica. Symp. Soc. Exp. Biol. 12, 139–163.
Estratto da Pubmed | Testo completo di Pubmed
Crick, F. (1970). Dogma centrale della biologia molecolare. Nature 227, 561–563.
Estratto su Pubmed | Testo completo di Pubmed
Hayden, E. C. (2011). La prova di RNA alterato suscita il dibattito. Nature 473, 432.
Estratto di Pubmed | Testo completo di Pubmed | Testo completo di CrossRef
Hekstra, D. R. e Leibler, S. (2012). Contingenza e leggi statistiche nella replica di ecosistemi microbici chiusi. Cella 149, 1164–1173.
Estratto di Pubmed | Testo completo di Pubmed | CrossRef Full Text
Kuwahara, H. e Schwartz, R. (2012).Guadagno stazionario stocastico in un processo di espressione genica con controllo della degradazione dellmRNA. J. R. Soc. Interfaccia 9, 1589–1598.
Estratto su Pubmed | Testo completo di Pubmed | CrossRef Full Text
Nie, L., Wu, G. e Zhang, W. (2006). Correlazione tra espressione di mRNA e abbondanza proteica influenzata da molteplici caratteristiche di sequenza correlate allefficienza traslazionale in Desulfovibrio vulgaris: unanalisi quantitativa. Genetica 174, 2229–2243.
Estratto su Pubmed | Testo completo di Pubmed | Testo completo di CrossRef
Prusiner, S. B. (1998). Prioni. Proc. Natl. Acad. Sci. USA 95, 13363–13383.
Estratto da Pubmed | Testo completo di Pubmed | Testo completo di CrossRef
Rosner, B. (2011). Fondamenti di biostatistica. 7th Edn. Boston, MA: Duxbury Press.
Selvarajoo, K. (2006). Scoperta del meccanismo di attivazione differenziale delle vie di segnalazione del recettore 4 tipo pedaggio nei knockout di MyD88. FEBS Lett. 580, 1457–1464.
Estratto di Pubmed | Testo completo di Pubmed | Testo completo di CrossRef
Selvarajoo, K. (2011). Legge macroscopica di conservazione rivelata nelle dinamiche di popolazione della segnalazione del recettore tipo pedaggio. Cell Commun. Segnale. 9, 9.
Estratto da Pubmed | Testo completo di Pubmed | Testo completo di CrossRef
Selvarajoo, K. (2012). Comprensione delle decisioni biologiche multimodali dalle dinamiche di singola cellula e popolazione. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 385–399.
Estratto su Pubmed | Testo completo di Pubmed | Testo completo di CrossRef
Stewart, T. R. (1990). Una scomposizione del coefficiente di correlazione e il suo utilizzo nellanalisi delle capacità di previsione. Previsioni del tempo. 5, 661–666.