Die Informationsverarbeitung ist in allen Bereichen der Wissenschaft von wesentlicher Bedeutung. In der Molekularbiologie ist das zentrale Dogma, das zuerst von Francis Crick (Crick, 1958, 1970) geprägt wurde, ein klassisches Rückgrat lebender Zellen, um Prozesse von der Zellteilung bis zum Tod über die DNA-, RNA- und Proteininformationswege grundlegend auszuführen. Insbesondere beschreibt das zentrale Dogma den Transfer von Sequenzinformationen während der DNA-Replikation, die Transkription in RNA und die Translation in Aminosäureketten, die Proteine bilden. Gleichzeitig heißt es auch, dass Informationen nicht von Protein zu Protein oder Nukleinsäure fließen können.
Seit dem Aufkommen systemischer Ansätze und Ansätze mit hohem Durchsatz in den letzten zwei Jahrzehnten umfassen diese umfassenden Schritte diese nicht komplexe regulatorische Details wurden einer intensiven Prüfung unterzogen. Die fehlenden regulatorischen Merkmale, wie die DNA-Korrektur- / Reparaturmechanismen und das alternative Spleißen von Prä-mRNA, führen mehrere Zwischenschritte ein. Diese zusätzlichen Schritte stören die Schlüsselschritte des Dogmas und verändern wahrscheinlich die Informationsdynamik. Darüber hinaus scheinen die Epigenetik oder die Rolle, die Chromatinstrukturen, DNA-Methylierung und Histonmodifikationen spielen, auch gegen die einfachen Wege des Dogmas zu verstoßen (Shapiro, 2009; Luco et al., 2011). Das in jüngster Zeit entdeckte Proteinspleißen oder die Fähigkeit eines Proteins (Inteine), seine eigene Sequenz zu verändern (Volkmann und Mootz, 2012), und Prionen, die andere Proteinsequenzen modifizieren (Prusiner, 1998), umgehen den Informationstransferweg des Dogma. Andere Untersuchungen berichteten über Fehler oder Fehlpaarungen zwischen RNA-Sequenzen und ihrer kodierenden DNA (Hayden, 2011; Li et al., 2011). Zusammengenommen werfen diese Daten Zweifel an der Gültigkeit des zentralen Dogmas im Kontext der heutigen Wissenschaft auf und stellen daher die Einfachheit des linearen Informationsflusses (DNA zu RNA und RNA zu Protein) in Frage.
Um die Dinge ins rechte Licht zu rücken, benötigen wir Analysewerkzeuge, die die Bedenken oder Diskrepanzen in Bezug auf die langjährige Theorie untersuchen. Eine einfache, aber äußerst nützliche Technik für die Suche nach globalen Eigenschaften in Datensätzen mit hohem Durchsatz ist die statistische Korrelationsanalyse, die weit verbreitet und erfolgreich zur Beobachtung von Mustern in komplexen Systemen wie Wetter (Stewart, 1990), Aktienmärkten (Lo und MacKinlay) eingesetzt wurde , 1988) und Cosmology (Amati et al., 2008). Es gibt verschiedene Arten von Korrelationsanalysen, die sowohl lineare (z. B. Pearson-Produktmoment) als auch nichtlineare (z. B. Spearmans Rang, gegenseitige Information) Abhängigkeiten bewerten (Steuer et al., 2002; Rosner, 2011) Die Pearson-Produkt-Moment-Korrelationsanalyse ist aufgrund ihrer Fähigkeit, die Organisationsstruktur in der einfachsten Form darzustellen, am beliebtesten geworden.
In der Biologie gab es zahlreiche Arbeiten, die die Korrelationen in der mRNA und untersuchten Proteinexpressionsdaten (siehe unten und Tabelle 1). Theoretisch liefern die Korrelationsanalysen beim Vergleich von zwei Proben, die hochdimensionale Daten (wie Microarray- und Proteomdaten) enthalten, ein Maß für die Abweichung von der Einheit als Quelle für Unterschiede zwischen den Proben Kurz gesagt, zwei Stichproben mit identischen und vollständig nicht identischen Informationen zeigen die Korrelation von Einheit (R2 = 1) bzw. Null (R2 = 0).
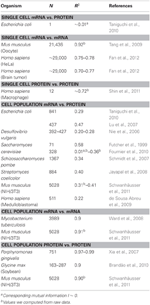
Tabelle 1. mRNA- und Protein-Ausdruck ession Korrelationen in verschiedenen Organismen.
Perfekte Korrelation (R2 = 1) ist eine idealisierte Situation, die technisch gesehen weit von der Realität entfernt ist oder experimentelles Rauschen allein stört und verringert die Korrelation. Darüber hinaus haben die letzten Jahre die Existenz von biologischem Rauschen hervorgehoben: Die Studien an einzelnen Zellen und Molekülen haben eine Stochastizität der Genexpressionsdynamik aufgrund des kombinatorischen Effekts niedriger molekularer Kopienzahlen und der quantalen Natur der Promotordynamik gezeigt (Raj und van Oudenaarden, 2009; Eldar und Elowitz, 2010). Andererseits zeigen klonale Populationen von Zellen zu jedem gemessenen Zeitpunkt eine Heterogenität in den Niveaus einer gegebenen Proteinexpression pro Zelle (Chang et al., 2008). Zusammen sind Stochastizität und Heterogenität wesentlich für die Diversifizierung des Zellschicksals, phänotypische Variationen und die Verstärkung intrazellulärer Signale (Locke et al., 2011; Selvarajoo, 2012).
Die stochastischen Schwankungen oder das intrinsische Rauschen verursachen Die Expression einer molekularen Spezies variiert zeitlich und zwischen den Zellen, was zu unkorrelierten Reaktionen führt (Elowitz et al., 2002). Dies gilt insbesondere für mRNAs und Proteine mit niedrigen Kopienzahlen. Somit kann die Korrelation zwischen Proben (Zellen) aufgrund von Eigenrauschen verringert werden (Fig. 1A). Andere Quellen für biologisches Rauschen aufgrund extrinsischer Faktoren sind Variabilität der Zellgröße, molekulare Kopienzahlen und Umgebungsschwankungen zwischen einzelnen Zellen.Diese Faktoren verzerren das deterministische zentrale Dogma und verändern wahrscheinlich starke Korrelationen in schwächere (Abbildung 1B).
Eine kürzlich durchgeführte Studie verglich die mRNA- und Proteinexpression von Escherichia coli zwischen einzelnen Zellen auf Einzelmolekülebene und lieferte ein Szenario, das das zentrale Dogma zutiefst in Frage stellt. Taniguchi et al. (2010) zeigten, dass es keine Korrelation (R2 ~ 0) zwischen einzelnen tufA-mRNA- und Proteinspiegeln in einzelnen Zellen gibt. Insbesondere kamen sie zu dem Schluss, dass der Mangel an Korrelation wahrscheinlich auf Unterschiede in der mRNA- und Proteinlebensdauer zurückzuführen ist. Obwohl dies eine plausible Erklärung ist, haben Taniguchi et al. achteten darauf, die lang anhaltende Hypothese nicht zu widerlegen, indem sie behaupteten, dass Zeitmittelwerte der mRNA-Spiegel mit den Proteinspiegeln korrelieren sollten. Es gab jedoch keine Beweise dafür, dass dies der tatsächliche Fall ist, und als wir nichtlineare Abhängigkeiten unter Verwendung gegenseitiger Informationen bewerteten (Steuer et al., 2002; Tsuchiya et al., 2010), bei Taniguchi et al. Wir fanden, dass das Ergebnis nicht abhängig ist, d. h. I ~ 0. Dies bestätigt, dass die Expression von mRNA zu Protein zwischen einzelnen Zellen auf Einzelmolekülebene eindeutig nicht miteinander zusammenhängt. Darüber hinaus ist beim Zoomen auf Einzelmolekülebene im Korrelationsdiagramm ersichtlich, dass ihre paarweisen Korrelationen schwach sind (1A, zur Veranschaulichung einfügen).
Insbesondere auf Zellpopulationsebene haben Taniguchi et al. konnten eine relativ hohe Korrelation zwischen mRNA- und Proteinexpression mit R2 = 0,29 zeigen (Abbildung 2A). Eine weitere unabhängige Studie von Lu et al. (2007) zeigten für die E. coli-Population ebenfalls eine relativ hohe Korrelation (R2 = 0,47). Ähnliche Analysen wurden an Saccharomyces cerevisiae (Futcher et al., 1999), murinem NIH / 3T3-Fibroblasten (Schwanhäusser et al., 2011) und mehreren anderen Zellpopulationen (Nie et al., 2006; Schmidt et al., 2007; Jayapal et al.) Durchgeführt al., 2008; de Sousa Abreu et al., 2009) zeigten alle korrelierte Strukturen zwischen transkriptomweiten und proteomweiten Ausdrücken (Tabelle 1). Warum gibt es also keine Korrelation zwischen einzelnen mRNA- und Proteinexpressionen in einzelnen Zellen, während auf Populationsebene kollektive Beziehungen zwischen mRNA- und Proteinexpressionen in großem Maßstab beobachtet werden?
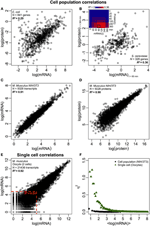
Abbildung 2. Omics-weite Ausdruckskorrelationen. Zellpopulationen: mRNA-Protein-Korrelationen in (A) E. coli (Taniguchi et al., 2010) und (B) S. cerevisiae (Fournier et al., 2010) zwischen mRNA-Expressionen bei t = 60 min und Proteinexpressionen bei t = 360 min. Einfügen: Die Korrelationsmatrix zwischen allen Zeitpunkten zeigt einen verzögerten Anstieg der Korrelationen zwischen mRNA und Proteinen. (C) mRNA- und (D) Proteinexpression zwischen zwei Proben von murinen NIH / 3T3-Zellen (Schwanhäusser et al., 2011). Einzelzellen: (E) mRNA-Expression zwischen zwei Oozyten (Tang et al., 2009). Die rot gepunkteten Linien zeigen die Regionen mit geringer mRNA-Expression an (log (mRNA) < 5). (F) Rauschen (η2) versus log (mRNA-Expression) für die Zellpopulation (NIH / 3T3, schwarze Punkte, Schwanhäusser et al., 2011) und einzelne Zellen (Oozyten, grüne Dreiecke, Tang et al., 2009). Jeder Punkt repräsentiert den Wert für eine Gruppe von P = 100 mRNAs. η2 ist für die Zellpopulation für alle mRNA-Expressionen nahe Null. Für einzelne Zellen ist η2 für mRNAs mit den niedrigsten Kopienzahlen am höchsten und für höhere Kopienzahlen gegen Null.
Wir glauben Es gibt zwei Hauptgründe für die Unterschiede. Erstens verringert, wie bereits erwähnt, Rauschen, ob biologischer oder nicht biologischer Natur, die Korrelation. Da Analysen an einzelnen Zellen die Bedeutung von Stochastizität und Variabilität gezeigt haben, sind diese Effekte entscheidend für die Verringerung der Korrelationen einzelner Zellen. Auf Ensemble-Ebene wird bei der Probenahme von Zellen in eine Population das gesamte (intrinsische + extrinsische) Rauschen reduziert, da sich das zufällige Rauschen über alle Bereiche molekularer Ausdrücke hinweg aufhebt (Abbildungen 1C – F), um die durchschnittliche Reaktion und Selbstorganisation zu ermitteln ( Karsenti, 2008; Selvarajoo, 2011; Hekstra und Leibler, 2012; Selvarajoo und Giuliani, 2012). Daher ergibt sich ein guter Grad an mRNA-Protein-Expressionskorrelation. Zweitens wurde für die Einzelzellstudie (Taniguchi et al., 2010) die individuelle mRNA-Protein-Expressionskorrelation über zahlreiche Zellen hinweg verglichen. In Zellpopulationsstudien wird der Vergleich jedoch vollständig über Tausende von mRNAs und Proteinen über mehrere Größenordnungen durchgeführt, die größer sind als der Expressionsbereich, der für ein einzelnes Molekül zwischen Zellen gefunden wurde. Dies führt daher zu höheren Korrelationen auf Populationsebene, da der Effekt einzelner molekularer Variationen vernachlässigbar wird.
Obwohl für Zellpopulationen korrelierte Strukturen beobachtet werden, gibt es konkrete Gründe für die große Abweichung von der perfekten Korrelation.Wie bereits erwähnt, ist ein wichtiger Punkt, dass mRNAs und Proteine nacheinander mit mehreren fehlenden Prozessen lokalisiert werden, die im zentralen Dogma nicht vertreten sind. Das Hinzufügen der fehlenden Zwischenprodukte entlang eines biochemischen Weges führt zu einer merklichen Verzögerung des Informationsflusses (Selvarajoo, 2006, 2011; Piras et al., 2011), und die Korrelation zwischen ihnen könnte darunter leiden. Dies könnte auch Teil der von Taniguchi et al. dass mRNA- und Proteinexpressionen unterschiedliche Lebensdauern haben. Bemerkenswerterweise wird diese Annahme in einer kürzlich durchgeführten Arbeit an mit Rapamycin behandelten S. cerevisiae gestützt, die zeigte, dass die zeitlichen Korrelationen der mRNA-Protein-Expression anfänglich niedrig waren, R2 = 0,01 nach 40 Minuten, jedoch über 360 Minuten nach der Störung die Korrelation zunahm. R2 = 0,36 (Fournier et al., 2010, 2B). Die Daten zeigen, dass bei chemischer Störung die anfängliche Reaktion zwischen mRNA- und Proteinexpression aufgrund von Zeitverzögerung und unterschiedlichen kinetischen Mechanismen zwischen ihnen sowie sekundären Effekten wie autokrinen oder parakrinen Signalstörungen abweicht (Shvartsman et al., 2002; Isalan et al., 2008). Wenn die Auswirkungen der Störung im Laufe der Zeit abgeschwächt werden, erfolgt die Wiederherstellung von Korrelationen.
Um die Annahme weiter zu überprüfen, dass sequentielle Verzögerungsprozesse oder unterschiedliche Lebensdauern für die Verringerung der mRNA-Protein-Korrelationen entscheidend sind, haben wir R2 zwischen den verglichen gleiche molekulare Spezies des zentralen Dogmas (z. B. zwischen mRNA und mRNA) in Zellpopulationen und Einzelzellen. Die transkriptomweite mRNA-mRNA-Expressionskorrelation zwischen Replikaten von NIH / 3T3 (Schwanhäusser et al., 2011) (2C) und Mycobacterium tuberculosis (Ward et al., 2008) Zellpopulationsproben ist beide sehr hoch, mit R2 > 0,9 (Tabelle 1). Solche starken Korrelationen werden auch zwischen Populationsproben für Protein-Protein-Expressionen in NIH / 3T3-Zellen (Schwanhäusser et al., 2011) (Abbildung 2D), Porphyromonas gingivalis (Xia et al., 2007) und Glycine max (Brandão et al. , 2010) (Tabelle 1). Da diese Daten, die dieselbe Spezies vergleichen, sehr hohe Korrelationen ergeben, ist es denkbar, dass die sequentiellen Verzögerungsprozesse oder unterschiedliche Lebensdauern für die Verringerung der Korrelationsstrukturen auf Populationsebene zwischen mRNA- und Proteinexpression verantwortlich sind.
In einzelnen murinen Oozyten ( Tang et al., 2009) wird beim Vergleich der gesamten mRNA-mRNA-Expression eine stark korrelierte Struktur beobachtet (R2 = 0,92, Abbildung 2E). Wenn man sich jedoch nur auf niedrig exprimierte mRNAs konzentriert (mit logarithmischen Ausdrücken < 5), senkt das stochastische Rauschen die paarweise Korrelation ziemlich dramatisch (R2 < 0,54). Um dieses Ergebnis zu untersuchen, bewerteten wir das Rauschen η2 = σ2XY / μ2XY über die gesamte mRNA-Expression (Abbildung 2F). Wir haben festgestellt, dass η2 für die niedrigsten Ausdrücke aufgrund des ausgeprägten Effekts stochastischer Schwankungen im Vergleich zu ihren Ausdrücken am höchsten ist und für höhere Ausdrücke gegen Null geht, wenn ein solches Rauschen weniger signifikant wird (Piras et al., 2012). Wie erwartet wird für die Zellpopulation über den gesamten Expressionsbereich ein Rauschen nahe Null beobachtet, da zufälliges Rauschen aufgehoben wird (Abbildungen 1E, F).
Es wurden auch stark korrelierte Strukturen für die gesamte mRNA-mRNA-Expression festgestellt berichtet für einzelne Krebszellen (Fan et al., 2012), wenn auch mit R2 ~ 0,7 weniger signifikant (Tabelle 1). Darüber hinaus zeigte der Vergleich der Protein-Protein-Expression in LPS-stimulierten menschlichen Makrophagen auch hohe Korrelationen, R2 ~ 0,72 (Shin et al., 2011) (Tabelle 1). Obwohl es keine Korrelation zwischen einzelnen mRNA-Protein-Expressionen in einzelnen Zellen gibt, ist die großräumige oder omikweite Korrelation zwischen denselben molekularen Spezies in einzelnen Zellen sehr hoch. Somit, ob einzelne Zellen oder Zellpopulationen Die omikweiten Daten zeigen, dass die Korrelationen zwischen derselben molekularen Spezies (mRNA vs. mRNA und Protein vs. Protein) deutlich höher sind als zwischen verschiedenen Spezies (mRNA vs. Protein). Dies spiegelt die Tatsache wider, dass Zeitverzögerungsprozesse und unterschiedliche Lebensdauern zwar der Schlüssel zur Verringerung von Korrelationen sind, diese Mechanismen jedoch nicht ausreichen, um den Mangel an Korrelationsstruktur zu unterstützen, der zwischen dem individuellen Transkript einzelner Zellen zu Proteinexpressionen beobachtet wird.
Also Bisher haben wir durch die Untersuchung der Expression von mRNAs und Proteinen verschiedener zellulärer Systeme in großem Maßstab gezeigt, dass Korrelationsstrukturen auf globaler Ebene entstehen. Die Korrelationsanalysen zeigen jedoch nur die Konnektivität zwischen zwei getesteten Proben und zeigen nicht die Richtung von Informationsfluss. Damit das zentrale Dogma auf globaler Ebene gültig ist, sollte der gesamte Informationsfluss von der DNA zu den Proteinen erfolgen. Ein solcher Informationsfluss wurde durch unzählige andere Studien nachgewiesen, bei denen die Rezeptoren von Zellpopulationen gestört und das Ergebnis überwacht werden Dynamik von Transkriptionsfaktoren, die an DNA binden, und Induktion von Genexpressionen in großem Maßstab (Abbildung 3A).Beispielsweise wurde im Fall von LPS-stimulierten Immunzellen gezeigt, dass die Aktivierung des Transkriptionsfaktors NF-κB nach etwa 15 Minuten erfolgt (Liu et al., 1999), die Induktion seiner nachgeschalteten Gene nach etwa 30 Minuten min (Liu et al., 1999; Xaus et al., 2000; Selvarajoo et al., 2008) und die Translation der entsprechenden Proteine im Bereich von 60–90 min (Kawai et al., 1999; Xaus et al ., 2000) (Fig. 3B). Eine solche sequentielle Richtung der gesamten Transkription zum Translationsinformationsfluss wird auch für Bakteriensysteme wie E. coli auf Zellpopulationsebene beobachtet (Golding et al., 2005).

Abbildung 3. Der Informationsfluss des zentralen Dogmas. (A) Schema der LPS / TLR4-induzierten TNF-Expression über den Transkriptionsfaktor NF-κB und das tnf-Gen nach linearem Informationsfluss. (B) Experimentelle zeitliche Profile der Promotorbindungsaktivität von NF-κB (obere Felder), tnf (mittlere Felder) und TNF (untere Felder) Expression auf Zellpopulationsebene. (C) Schematische zeitliche Profile der Promotordynamik, mRNA- und Proteinexpression auf Einzelzellenebene (Raj und van Oudenaarden, 2009).
Alternativ zeigen Untersuchungen bei Einzelzellauflösung zufällige Schwankungen über den linearen Informationsfluss: Die Transkriptionsfaktoren, die an DNA-Promotorregionen binden, sind quantal, was zu einem Berstverhalten der mRNA-Transkription führt und anschließend eine Variabilität in der Proteintranslation induziert. sogar zwischen identischen Zellen (3C) (Raj und van Oudenaarden, 2009; Eldar und Elowitz, 2010; Locke et al., 2011; Hekstra und Leibler, 2012; Selvarajoo, 2012). Infolgedessen ist die individuelle molekulare Reaktion für einzelne Zellen zu einem bestimmten Zeitpunkt im Vergleich zur Populationsdurchschnittsskala ziemlich verrauscht (Selvarajoo, 2011).
Schlussfolgerungen
Die in In diesem Artikel werden die Unterschiede in der Reihenfolge der Korrelationswerte hervorgehoben, die zwischen Arten im zentralen Dogma gegenüber Zellpopulationen und Einzelzellen beobachtet wurden. Die statistischen Analysen von Zellpopulationen zeigen, dass die Expressionskorrelation zwischen derselben molekularen Spezies sehr hoch und zwischen Spezies mäßig hoch ist. Obwohl Einzelzellkorrelationen zwischen derselben Spezies mit Zellpopulationen vergleichbar sind, zeigten sie aufgrund der ausgeprägten Wirkung von biologischem Rauschen eine größere Streuung in ihren Expressionskurven, insbesondere bei Transkripten mit niedrigen Kopienzahlen. Insbesondere wird die paarweise Korrelation einzelner Zellen für einzelne Moleküle zu Null (Taniguchi et al., 2010). Tatsächlich ist bekannt, dass stochastische Fluktuationen und Variabilität der molekularen Expressionen bei der Erzeugung der Entscheidung über das Zellschicksal und beim Tippen auf zelluläre Zustände eine Funktion haben (Losick) und Desplan, 2008; Eldar und Elowitz, 2010; Kuwahara und Schwartz, 2012). Wir glauben, dass die starken omikweiten Korrelationen auf enge Gen- und Proteinregulationsnetzwerke über Tausende von Molekülen zurückzuführen sind (Barabási und Oltvai, 2004; Karsenti) , 2008), was zu emergenten Durchschnittsreaktionen führt. Bei der Analyse einer kleinen Anzahl oder einzelner Moleküle kann die Korrelationsstruktur nicht beobachtet werden.
Insgesamt ist es denkbar, dass die Betrachtung des Informationsflusses von einzelner DNA zu Protein das zentrale Dogma in Frage stellt Da die Reaktion jedes Moleküls zu einem bestimmten Zeitpunkt wahrscheinlich nicht korreliert, deutet die Beobachtung der durchschnittlichen deterministischen Reaktion global jedoch darauf hin, dass das Nettogleichgewicht des Gens Die Informationen bleiben ganz rechts von den Pfaden. Daher sollte das zentrale Dogma als makroskopischer zellulärer Informationsfluss in omikweitem Maßstab und nicht auf der Ebene einzelner Gene bis Proteine betrachtet werden. Insofern glauben wir, dass seine Einfachheit weiterhin eine der einflussreichsten theoretischen Säulen lebender Systeme bleiben wird.
Erklärung zu Interessenkonflikten
Die Autoren erklären, dass die Forschung in durchgeführt wurde das Fehlen jeglicher kommerzieller oder finanzieller Beziehungen, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagung
Kentaro Hayashi wird für Kommentare gedankt. Der Forschungsfonds der Stadt Tsuruoka und der Präfektur Yamagata wird für ihre Unterstützung geschätzt.
Crick, F. (1958). Zur Proteinsynthese. Symp. Soc. Exp. Biol. 12, 139–163.
Pubmed Abstract | Pubmed-Volltext
Crick, F. (1970). Zentrales Dogma der Molekularbiologie. Nature 227, 561–563.
Pubmed Abstract | Veröffentlichter Volltext
Hayden, E. C. (2011). Hinweise auf veränderte RNA regen die Debatte an. Nature 473, 432.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Hekstra, D. R. und Leibler, S. (2012). Kontingenz- und statistische Gesetze in replizierten mikrobiellen geschlossenen Ökosystemen. Cell 149, 1164–1173.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Kuwahara, H. und Schwartz, R. (2012).Stochastischer Steady-State-Gewinn in einem Genexpressionsprozess mit mRNA-Abbaukontrolle. J. R. Soc. Interface 9, 1589–1598.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Nie, L., Wu, G. und Zhang, W. (2006). Korrelation der mRNA-Expression und der Proteinhäufigkeit, die durch mehrere Sequenzmerkmale im Zusammenhang mit der Translationseffizienz bei Desulfovibrio vulgaris beeinflusst wird: eine quantitative Analyse. Genetics 174, 2229–2243.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Prusiner, S. B. (1998). Prionen. Proc. Natl. Acad. Sci. USA 95, 13363–13383.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Rosner, B. (2011). Grundlagen der Biostatistik. 7. Aufl. Boston, MA: Duxbury Press.
Selvarajoo, K. (2006). Entdeckung der differentiellen Aktivierungsmaschinerie der Toll-like-Rezeptor-4-Signalwege bei MyD88-Knockouts. FEBS Lett. 580, 1457–1464.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Selvarajoo, K. (2011). Das makroskopische Erhaltungsgesetz zeigt sich in der Populationsdynamik der mautähnlichen Rezeptorsignalisierung. Cell Commun. Signal. 9, 9.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Selvarajoo, K. (2012). Verständnis multimodaler biologischer Entscheidungen aus der Dynamik einzelner Zellen und Populationen. Wiley Interdiscip. Rev. Syst. Biol. Med. 4, 385–399.
Pubmed Abstract | Pubmed Volltext | CrossRef-Volltext
Stewart, T. R. (1990). Eine Zerlegung des Korrelationskoeffizienten und seine Verwendung bei der Analyse der Prognosefähigkeit. Wettervorhersage. 5, 661–666.